Riepilogo Dallo scoppio della malattia da coronavirus 2019 (COVID-19), i medici hanno fatto ogni sforzo per comprendere la malattia ed è stato identificato un breve ritratto delle sue caratteristiche cliniche. Nella pratica clinica, abbiamo notato che molti pazienti affetti da COVID-19 gravi o critici hanno sviluppato manifestazioni cliniche tipiche dello shock , tra cui estremità fredde e polsi periferici deboli, anche in assenza di ipotensione palese. Comprendere il meccanismo della sepsi virale nel COVID-19 è la garanzia per esplorare una migliore assistenza clinica per questi pazienti. Con le prove raccolte dagli studi autoptici su COVID-19 e dalla ricerca scientifica di base sulla sindrome respiratoria acuta grave coronavirus 2 (SARS-CoV-2) e SARS-CoV, abbiamo avanzato diverse ipotesi sulla patogenesi della SARS-CoV. -2 dopo molteplici cicli di discussione tra ricercatori di scienze di base, patologi e medici che lavorano su COVID-19. Ipotizziamo che un processo chiamato sepsi virale sia cruciale per il meccanismo della malattia di COVID-19. Sebbene queste idee possano rivelarsi imperfette o addirittura errate in seguito, riteniamo che possano fornire input e indicazioni per la ricerca di base in questo momento. |
L’epidemia della sindrome respiratoria acuta grave coronavirus 2 (SARS-CoV-2), segnalata per la prima volta a Wuhan, in Cina, nel dicembre 2019, ha avuto un enorme impatto sulla Cina e sul mondo intero. La malattia causata dalla SARS-CoV-2 è chiamata malattia da coronavirus 2019 (COVID-19). Entro il 19 marzo 2020, il numero di casi confermati era aumentato a oltre 200.000. Sebbene la maggior parte dei pazienti infettati da SARS-CoV-2 presentasse una malattia lieve, circa il 5% dei pazienti presentava lesioni polmonari gravi o addirittura disfunzioni multiorgano , con un tasso di mortalità dell’1,4%1.
In pratica, abbiamo notato che molti pazienti affetti da COVID-19 gravi o critici hanno sviluppato manifestazioni cliniche tipiche dello shock, tra cui estremità fredde e polsi periferici deboli, anche in assenza di ipotensione palese. Molti di questi pazienti presentavano una grave acidosi metabolica , indice di una possibile disfunzione del microcircolo. Inoltre, alcuni pazienti presentavano insufficienza epatica e renale oltre a gravi lesioni polmonari.
Questi pazienti soddisfacevano i criteri diagnostici per sepsi e shock settico secondo il Sepsi-3 International Consensus 3 ma l’infezione da SARS-CoV-2 sembrava essere l’unica causa nella maggior parte di essi.1 Sono state effettuate colture di campioni di sangue e del tratto respiratorio inferiore risultato negativo per batteri e funghi nel 76% dei pazienti con sepsi in una coorte COVID-19.
Pertanto, la sepsi virale sarebbe più accurata per descrivere le manifestazioni cliniche di pazienti affetti da COVID-19 gravi o critici.5 Comprendere il meccanismo della sepsi virale in COVID-19 è la garanzia per esplorare una migliore assistenza clinica per questi pazienti.
Infezione da virus e patogenesi del COVID-19 negli organi
Negli studi di biopsia o autopsia, la patologia polmonare sia per i pazienti con COVID-19 in fase precoce6 che per quella tardiva7 ha mostrato un danno alveolare diffuso con la formazione di membrane ialine, cellule mononucleari e macrofagi che infiltrano gli spazi aerei e un ispessimento diffuso della parete alveolare. Particelle virali sono state osservate nelle cellule epiteliali alveolari bronchiali di tipo 2 mediante microscopia elettronica8, 9.
Inoltre, in alcuni pazienti erano presenti atrofia della milza, necrosi dei linfonodi ilari, emorragia focale nel rene, ingrossamento del fegato con infiltrazione di cellule infiammatorie, edema e degenerazione diffusa dei neuroni nel cervello 8, 9.
Particelle infettive del virus SARS-CoV-2 sono state isolate da campioni respiratori,10 così come da campioni fecali11 e urina (Zhao J, Guangzhou Medical University, comunicazione personale) di pazienti con COVID-19, suggerendo che la disfunzione Danno multiorgano nella grave COVID19 è causato almeno in parte da un attacco diretto del virus. Tuttavia, al momento non ci sono rapporti sulle osservazioni post-mortem della diffusa diffusione di particelle virali tramite autopsia.
Se la SARS-CoV-2 possa colpire direttamente organi diversi dal polmone, in particolare quegli organi con un’elevata espressione dell’enzima 2 di conversione dell’angiotensina (ACE2) 12,13 e gli organi con L-SIGN(14) come possibili recettori cellulari alternativi per la SARS- CoV-2, deve essere studiato ulteriormente. Inoltre, la questione di come la SARS-CoV-2 si diffonde agli organi extra-polmonari rimane un enigma. È stata osservata una variazione genomica del SARS-CoV-2 circolante e la differenza nella virulenza necessita di ulteriori indagini15.
Risposta immunitaria alla SARS-CoV-2 e alla sepsi virale
È stato dimostrato che le citochine e le chemochine proinfiammatorie includono il fattore di necrosi tumorale (TNF) α, l’interleuchina 1β (IL-1β), IL-6, il fattore stimolante le colonie di granulociti, la proteina 10 indotta dall’interferone gamma, la proteina chemoattrattiva Proteine infiammatorie monocitarie e macrofagiche 1 -α erano significativamente elevati nei pazienti con COVID-19, 16, 17. Come nell’infezione influenzale grave, la tempesta di citochine potrebbe svolgere un ruolo importante nell’immunopatologia di COVID-19. .
Precedenti studi hanno rivelato che le cellule epiteliali polmonari, i macrofagi e le cellule dendritiche esprimono tutte citochine in una certa misura durante l’infezione influenzale attraverso l’attivazione di recettori di riconoscimento del pattern (compresi i recettori Toll-like TLR3, TLR7 e TLR8), del gene I inducibile dall’acido retinoico e dei membri della famiglia dei recettori NOD18-simili.
Tuttavia, al momento si sa poco sulla situazione relativa al COVID-19. È fondamentale identificare la fonte primaria della tempesta di citochine in risposta all’infezione da SARS-CoV-2 e i meccanismi virologici dietro la tempesta di citochine. Sarebbe anche rilevante chiarire la cinetica dell’attivazione delle citochine durante l’infezione da SARS-CoV-2: quando furono rilasciate le prime citochine e quali erano?
Inoltre, resta da affrontare se il danno tissutale diretto indotto dal virus, la tempesta sistemica di citochine o gli effetti sinergici di entrambi contribuiscono alla disfunzione multiorgano dei pazienti gravi con COVID-19.
Inoltre, vale la pena monitorare se il blocco di uno di questi mediatori proinfiammatori possa influenzare l’esito clinico. Sono stati proposti anticorpi monoclonali anti-IL-6R o corticosteroidi per alleviare la risposta infiammatoria.
Tuttavia, l’IL-6 potrebbe svolgere un ruolo importante nell’avviare una risposta preliminare contro l’infezione virale promuovendo la clearance virale mediata dai neutrofili, poiché uno studio ha rivelato che la carenza di IL-6 o IL-6R ha portato alla persistenza dell’infezione influenzale e infine alla morte. nei topi.19 E l’uso dei corticosteroidi rimane controverso20, 21.
Tuttavia, la risposta immunitaria disregolata presenta anche uno stadio di soppressione immunitaria dopo la fase proinfiammatoria. È caratterizzata da una riduzione sostenuta e sostanziale della conta dei linfociti periferici, principalmente delle cellule T CD4 e CD8 nei pazienti con COVID-19, ed è associata ad un alto rischio di sviluppare un’infezione batterica secondaria. Questa condizione, nota come linfopenia , è stata riscontrata anche nell’influenza grave e in altre infezioni virali respiratorie.
È stato dimostrato che il grado di linfopenia è correlato alla gravità di COVID-19.17
Il meccanismo alla base della linfopenia rimane sconosciuto. Precedenti studi hanno dimostrato che particelle virali simili alla SARS e RNA di SARS-CoV sono stati rilevati nei linfociti T isolati da campioni di sangue periferico, milza, linfonodi e tessuto linfoide di vari organi 22,23 suggerendo che la SARS-CoV potrebbe infettare i T cellule direttamente . I domini di legame dei recettori delle proteine spike tra SARS-CoV-2 e SARS-CoV mostrano un elevato grado di coerenza 24. 25 e l’RNA di SARS-CoV-2 è stato rilevato anche nei campioni di sangue16.
Pertanto, è ragionevole supporre che, oltre alla morte cellulare indotta dall’attivazione indotta dall’interazione di Fas e del ligando di Fas, nonché dall’asse del ligando che induce l’apoptosi correlata al TNF, 26
SARS-CoV-2 potrebbe infettare direttamente i linfociti , in particolare le cellule T, e avviano o promuovono la morte delle cellule linfocitarie, portando infine a linfopenia e risposte antivirali compromesse. Tuttavia tale ipotesi necessita di essere approfondita.
Occorre inoltre identificare quali tipi di morte cellulare si verificano nei linfociti dopo l’infezione da SARS-CoV-2. Inoltre, è interessante il fatto che i linfociti manchino di espressione di ACE2, suggerendo un meccanismo alternativo attraverso il quale SARS-CoV-2 impegna i linfociti T.13 Se i macrofagi alveolari possano o meno fagocitare particelle virali e poi trasferirle ai linfociti è una questione aperta nel campo.
COVID-19 e coagulazione anormale
Gli studi hanno rivelato che il 71,4% dei non sopravvissuti al COVID-19 ha raggiunto il grado di coagulazione intravascolare disseminata a cielo aperto (≥5 punti secondo i criteri della International Society on Thrombosis and Haemostasis) 27 e ha mostrato risultati di coagulazione anormali durante le fasi. più tardi della malattia; Concentrazioni particolarmente elevate di D-dimero e di altri prodotti di degradazione della fibrina erano significativamente associate a una prognosi sfavorevole4.
Tuttavia, i meccanismi specifici della coagulopatia non sono stati ancora identificati. Resta da esplorare se SARS-CoV-2 sia in grado di attaccare direttamente le cellule endoteliali vascolari che esprimono alti livelli di ACE2,13
e quindi portare a coagulazione anormale e sepsi.
Nel frattempo, ACE2 è anche un importante regolatore della pressione sanguigna . Un’elevata espressione di ACE2 nel sistema circolatorio dopo l’infezione da SARS-CoV-2 potrebbe contribuire parzialmente all’ipotensione settica .
Sono state sollevate domande sull’uso del bloccante del recettore dell’angiotensina II (ARB) e della terapia con ACE inibitori per i pazienti con ipertensione COVID-19.
Alcuni ricercatori hanno suggerito che gli ACE inibitori potrebbero apportare benefici a questi pazienti riducendo l’infiammazione polmonare 29 mentre altri hanno sostenuto che gli ACE inibitori potrebbero migliorare l’ingresso virale regolando i livelli di ACE 230. Tuttavia, vi sono poche evidenze cliniche sul rischio del trattamento dei pazienti affetti da COVID-19 con ARB o ACE inibitori. Sono necessarie ulteriori ricerche per esplorare se questi farmaci inibiscono o favoriscono l’ingresso virale.
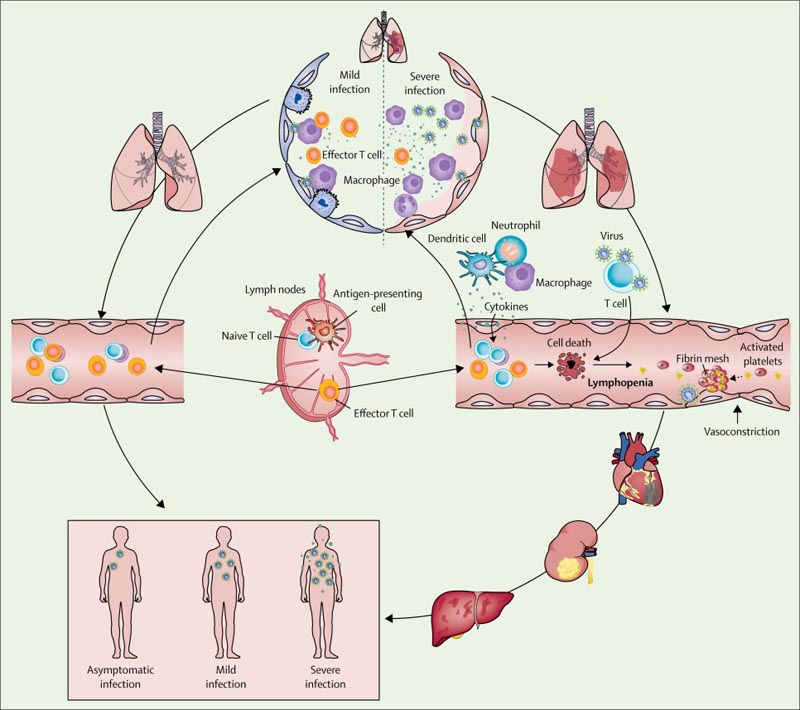 Figura Presenza ed evoluzione della sindrome respiratoria acuta grave, sepsi virale da coronavirus 2
Figura Presenza ed evoluzione della sindrome respiratoria acuta grave, sepsi virale da coronavirus 2
Conclusioni
Sulla base delle osservazioni di pazienti con COVID-19, abbiamo ipotizzato che, nei casi lievi , i macrofagi residenti che avviano le risposte infiammatorie polmonari fossero in grado di contenere il virus dopo l’infezione da SARS-CoV-2; Sia la risposta immunitaria innata che quella adattativa sono state stabilite in modo efficiente per frenare la replicazione virale in modo che il paziente si riprendesse rapidamente.
Tuttavia, nei casi gravi o critici di COVID-19, l’integrità della barriera epiteliale-endoteliale (aria-sangue) è stata gravemente compromessa. Oltre alle cellule epiteliali, SARS-CoV-2 può attaccare anche le cellule endoteliali dei capillari polmonari, provocando una grande quantità di essudato di componenti del plasma nella cavità alveolare.
In risposta all’infezione da SARS-CoV-2, i macrofagi alveolari o le cellule epiteliali potrebbero produrre varie citochine e chemochine proinfiammatorie. A seguito di questo cambiamento, i monociti e i neutrofili sono stati chemiotattici nel sito dell’infezione per eliminare questi essudati con particelle virali e cellule infette, provocando un’infiammazione incontrollata .
In questo processo, a causa della sostanziale riduzione e disfunzione dei linfociti, la risposta immunitaria adattativa non può essere avviata in modo efficace. L’infezione virale incontrollata porta ad un aumento dell’infiltrazione di macrofagi e ad un ulteriore peggioramento del danno polmonare.
Nel frattempo, l’attacco diretto ad altri organi da parte del SARS-CoV-2 disseminato, la patogenesi immunitaria causata dalla tempesta sistemica di citochine e le disfunzioni della microcircolazione portano insieme alla sepsi virale . Pertanto, una terapia antivirale efficace e misure per modulare la risposta immunitaria innata e ripristinare la risposta immunitaria adattativa sono essenziali per spezzare il circolo vizioso e migliorare i risultati dei pazienti.
Dallo scoppio del COVID-19, i medici hanno fatto del loro meglio per comprendere la malattia ed è stato identificato un breve ritratto delle sue caratteristiche cliniche. Tuttavia, ci sono ancora domande aperte sui meccanismi delle osservazioni.
Con le prove raccolte dagli studi autoptici sul COVID-19 e dalla ricerca scientifica di base su SARS-CoV-2 e SARS-CoV, abbiamo avanzato diverse ipotesi sulla patogenesi del SARS-CoV-2 dopo molteplici cicli di discussione tra i ricercatori. di scienze di base, patologi e medici che lavorano su COVID-19.
Ipotizziamo che un processo chiamato sepsi virale sia cruciale per il meccanismo della malattia di COVID-19. Anche se queste idee potrebbero rivelarsi imperfette o addirittura errate in seguito, riteniamo che sollevino interrogativi per la ricerca futura.
- Sono necessarie future ricerche scientifiche per esplorare se la SARS-CoV-2 attacca direttamente le cellule endoteliali vascolari e per esaminare l’effetto della SARS-CoV-2 sulla coagulazione e sulla diffusione del virus.
- Dovrebbero essere eseguiti studi clinici ed esperimenti sugli animali per valutare l’effetto degli ARB e degli ACE inibitori sull’esito dell’infezione da SARS-CoV-2 in vivo.
- Dovrebbero essere compiuti sforzi per confermare se SARS-CoV-2 infetta direttamente i linfociti e come influenza la risposta immunitaria adattativa.
- Anche la cinetica della risposta delle citochine durante l’infezione da SARS-CoV-2 necessita di ulteriori indagini.
- L’efficacia delle terapie immunomodulatorie dovrebbe essere valutata in studi clinici randomizzati.















