Zusammenfassung Seit dem Ausbruch der Coronavirus-Krankheit 2019 (COVID-19) haben Ärzte alle Anstrengungen unternommen, um die Krankheit zu verstehen, und es wurde ein kurzes Porträt ihrer klinischen Merkmale erstellt. In der klinischen Praxis haben wir festgestellt, dass viele schwer oder kritisch erkrankte COVID-19-Patienten typische klinische Schocksymptome entwickelten , einschließlich kalter Extremitäten und schwacher peripherer Pulse, selbst wenn keine offensichtliche Hypotonie vorlag. Das Verständnis des Mechanismus der Virussepsis bei COVID-19 ist die Garantie für eine bessere klinische Versorgung dieser Patienten. Mit Beweisen aus Autopsiestudien zu COVID-19 und wissenschaftlicher Grundlagenforschung zum schweren akuten respiratorischen Syndrom Coronavirus 2 (SARS-CoV-2) und SARS-CoV haben wir mehrere Hypothesen zur Pathogenese von SARS-CoV aufgestellt. -2 nach mehreren Diskussionsrunden zwischen Grundlagenforschern, Pathologen und Klinikern, die an COVID-19 arbeiten. Wir gehen davon aus, dass ein Prozess namens Virussepsis entscheidend für den Krankheitsmechanismus von COVID-19 ist. Auch wenn sich diese Ideen später möglicherweise als unvollkommen oder sogar falsch erweisen, glauben wir, dass sie der Grundlagenforschung zu diesem Zeitpunkt Input und Orientierung geben können. |
Einführung
Der Ausbruch des schweren akuten respiratorischen Syndroms Coronavirus 2 (SARS-CoV-2), der erstmals im Dezember 2019 in Wuhan, China, gemeldet wurde, hatte enorme Auswirkungen auf China und die ganze Welt. Die durch SARS-CoV-2 verursachte Krankheit wird als Coronavirus-Krankheit 2019 (COVID-19) bezeichnet. Bis zum 19. März 2020 stieg die Zahl der bestätigten Fälle auf über 200.000. Obwohl die meisten mit SARS-CoV-2 infizierten Patienten eine leichte Erkrankung hatten, erlitten etwa 5 % der Patienten eine schwere Lungenschädigung oder sogar eine Funktionsstörung mehrerer Organe , was zu einer Sterblichkeitsrate von 1,4 % führte1.
In der Praxis stellten wir fest, dass viele schwer oder kritisch erkrankte COVID-19-Patienten typische klinische Schocksymptome entwickelten, darunter kalte Extremitäten und schwache periphere Pulse, selbst wenn keine offensichtliche Hypotonie vorlag. Viele dieser Patienten zeigten eine schwere metabolische Azidose , was auf eine mögliche Störung der Mikrozirkulation hindeutet. Darüber hinaus litten einige Patienten zusätzlich zu einer schweren Lungenschädigung an Leber2- und Nierenversagen.
Diese Patienten erfüllten die diagnostischen Kriterien für Sepsis und septischen Schock gemäß dem Sepsis-3 International Consensus 3, bei den meisten von ihnen schien jedoch eine SARS-CoV-2-Infektion die einzige Ursache zu sein.1 Kulturen von Blutproben und der unteren Atemwege wurden erstellt bei 76 % der Patienten mit Sepsis in einer COVID-19-Kohorte als negativ für Bakterien und Pilze befunden.
Daher würde Virussepsis die klinischen Manifestationen schwerer oder kritischer COVID-19-Patienten genauer beschreiben.5 Das Verständnis des Mechanismus der Virussepsis bei COVID-19 ist die Garantie für die Erforschung einer besseren klinischen Versorgung dieser Patienten.
Virusinfektion und Pathogenese von COVID-19 in Organen
In Biopsie- oder Autopsiestudien zeigte die Lungenpathologie sowohl bei COVID-19-Patienten in der frühen6 als auch in der späten Phase7 eine diffuse Alveolarschädigung mit der Bildung hyaliner Membranen, mononukleären Zellen und Makrophagen, die Lufträume infiltrieren, sowie eine diffuse Verdickung der Alveolarwand. Durch Elektronenmikroskopie wurden Viruspartikel in bronchialen Alveolarepithelzellen vom Typ 2 beobachtet8, 9.
Darüber hinaus traten bei einigen Patienten eine Milzatrophie, eine Nekrose der Hiluslymphknoten, eine fokale Blutung in der Niere, eine vergrößerte Leber mit Infiltration von Entzündungszellen, Ödeme und eine vereinzelte Degeneration von Neuronen im Gehirn auf 8, 9.
Infektiöse SARS-CoV-2-Viruspartikel wurden aus Atemwegsproben10 sowie Stuhlproben11 und Urin (Zhao J, Medizinische Universität Guangzhou, persönliche Mitteilung) von Patienten mit COVID-19 isoliert, was darauf hindeutet, dass bei schwerem COVID-19 eine Funktionsstörung mehrerer Organe vorliegt wird zumindest teilweise durch einen direkten Angriff des Virus verursacht. Derzeit liegen jedoch keine Berichte über postmortale Beobachtungen der weitverbreiteten Verbreitung von Viruspartikeln durch Autopsie vor.
Ob SARS-CoV-2 direkt auf andere Organe als die Lunge abzielen kann, insbesondere auf Organe mit hoher Expression des Angiotensin-Converting-Enzyms 2 (ACE2) 12,13 und Organe mit L-SIGN(14) als mögliche alternative zelluläre Rezeptoren für SARS- CoV-2, es muss weiter untersucht werden. Darüber hinaus bleibt die Frage, wie sich SARS-CoV-2 auf extrapulmonale Organe ausbreitet, ein Rätsel. Es wurde eine genomische Variation des zirkulierenden SARS-CoV-2 beobachtet, und der Unterschied in der Virulenz bedarf weiterer Untersuchungen15.
Immunantwort auf SARS-CoV-2 und Virussepsis
Zu den proinflammatorischen Zytokinen und Chemokinen zählen nachweislich Tumornekrosefaktor (TNF) α, Interleukin 1β (IL-1β), IL-6, Granulozytenkolonie-stimulierender Faktor, Interferon-Gamma-induziertes Protein 10, chemoattraktives Protein Monozyten und Makrophagen-Entzündungsproteine 1 -α waren bei Patienten mit COVID-19 signifikant erhöht, 16, 17. Wie bei einer schweren Influenza-Infektion könnte der Zytokinsturm eine wichtige Rolle bei der Immunpathologie von COVID-19 spielen. .
Frühere Studien zeigten, dass Lungenepithelzellen, Makrophagen und dendritische Zellen alle während einer Influenza-Infektion in gewissem Maße Zytokine exprimieren, und zwar durch die Aktivierung von Mustererkennungsrezeptoren (einschließlich der Toll-like-Rezeptoren TLR3, TLR7 und TLR8), des Retinsäure-induzierbaren Gens I und seiner Mitglieder der NOD18-ähnlichen Rezeptorfamilie.
Derzeit ist jedoch wenig über die Situation im Zusammenhang mit COVID-19 bekannt. Es ist von entscheidender Bedeutung, die primäre Quelle des Zytokinsturms als Reaktion auf eine SARS-CoV-2-Infektion und die virologischen Mechanismen hinter dem Zytokinsturm zu identifizieren. Es wäre auch relevant, die Kinetik der Zytokinaktivierung während einer SARS-CoV-2-Infektion aufzuklären: Wann wurden die ersten Zytokine freigesetzt und was waren sie?
Darüber hinaus muss noch geklärt werden, ob eine direkte virusinduzierte Gewebeschädigung, ein systemischer Zytokinsturm oder synergistische Effekte beider zur Multiorganfunktionsstörung schwerer COVID-19-Patienten beitragen.
Darüber hinaus lohnt es sich zu überwachen, ob die Blockierung eines dieser proinflammatorischen Mediatoren das klinische Ergebnis beeinflussen würde. Zur Linderung der Entzündungsreaktion wurden monoklonale Anti-IL-6R-Antikörper oder Kortikosteroide vorgeschlagen.
Allerdings könnte IL-6 eine wichtige Rolle bei der Auslösung einer vorläufigen Reaktion gegen eine Virusinfektion spielen, indem es die durch Neutrophile vermittelte Virusclearance fördert, da eine Studie ergab, dass ein Mangel an IL-6 oder IL-6R zum Fortbestehen der Influenzainfektion und schließlich zum Tod führte bei Mäusen.19 Und der Einsatz von Kortikosteroiden bleibt umstritten20, 21.
Allerdings gibt es bei der fehlregulierten Immunantwort nach der proinflammatorischen Phase auch ein Stadium der Immunsuppression . Es zeichnet sich durch eine anhaltende und erhebliche Verringerung der peripheren Lymphozytenzahl , hauptsächlich CD4- und CD8-T-Zellen, bei Patienten mit COVID-19 aus und ist mit einem hohen Risiko für die Entwicklung einer sekundären bakteriellen Infektion verbunden. Dieser als Lymphopenie bekannte Zustand wurde auch bei schwerer Grippe und anderen Virusinfektionen der Atemwege gefunden.
Es wurde gezeigt, dass der Grad der Lymphopenie mit der Schwere von COVID-19 korreliert.17
Der Mechanismus, der der Lymphopenie zugrunde liegt, ist weiterhin unbekannt. Frühere Studien haben gezeigt, dass SARS-ähnliche Viruspartikel und SARS-CoV-RNA in T-Lymphozyten nachgewiesen wurden, die aus Proben von peripherem Blut, Milz, Lymphknoten und Lymphgewebe verschiedener Organe isoliert wurden 22,23, was darauf hindeutet, dass SARS-CoV T. infizieren könnte Zellen direkt . Die Rezeptorbindungsdomänen der Spike-Proteine zwischen SARS-CoV-2 und SARS-CoV weisen einen hohen Grad an Konsistenz auf24.25 und SARS-CoV-2-RNA wurde auch in Blutproben nachgewiesen16.
Daher ist es vernünftig anzunehmen, dass zusätzlich zu dem aktivierungsinduzierten Zelltod, der durch Fas- und Fas-Ligand-Wechselwirkung induziert wird, sowie der TNF-bedingten Apoptose-induzierenden Ligandenachse 26
SARS-CoV-2 Lymphozyten direkt infizieren könnte . insbesondere T-Zellen, und initiieren oder fördern den Zelltod von Lymphozyten, was schließlich zu Lymphopenie und beeinträchtigten antiviralen Reaktionen führt. Allerdings muss eine solche Hypothese weiter untersucht werden.
Es muss auch ermittelt werden, welche Arten des Zelltods in Lymphozyten nach einer SARS-CoV-2-Infektion auftreten. Darüber hinaus ist es interessant, dass Lymphozyten keine ACE2-Expression aufweisen, was auf einen alternativen Mechanismus hindeutet, durch den SARS-CoV-2 T-Lymphozyten angreift.13 Ob Alveolarmakrophagen Viruspartikel phagozytieren und sie dann auf Lymphozyten übertragen können, ist auf diesem Gebiet eine offene Frage.
COVID-19 und abnormale Gerinnung
Studien haben ergeben, dass 71,4 % der COVID-19-Nichtüberlebenden den Grad der offenen disseminierten intravaskulären Gerinnung erreichten (≥5 Punkte gemäß den Kriterien der International Society on Thrombosis and Haemostasis) 27 und während der Stadien abnormale Gerinnungsergebnisse zeigten. später der Krankheit; Insbesondere erhöhte Konzentrationen von D-Dimer und anderen Fibrinabbauprodukten waren signifikant mit einer schlechten Prognose verbunden4.
Die spezifischen Mechanismen der Koagulopathie sind jedoch noch nicht identifiziert. Ob SARS-CoV-2 in der Lage ist, vaskuläre Endothelzellen, die hohe Mengen an ACE2,13 exprimieren, direkt anzugreifen
und dann zu abnormaler Gerinnung und Sepsis zu führen, muss noch untersucht werden.
Mittlerweile ist ACE2 auch ein wichtiger Blutdruckregulator . Eine hohe Expression von ACE2 im Kreislaufsystem nach einer SARS-CoV-2-Infektion könnte teilweise zur septischen Hypotonie beitragen .
Es wurden Fragen zur Anwendung von Angiotensin-II-Rezeptorblockern (ARB) und einer ACE-Hemmer- Therapie bei Patienten mit COVID-19-Bluthochdruck aufgeworfen.
Einige Forscher schlugen vor, dass ACE-Hemmer diesen Patienten zugute kommen könnten, indem sie Lungenentzündungen reduzieren 29, während andere argumentierten, dass ACE-Hemmer den Viruseintritt verbessern könnten, indem sie den ACE-Spiegel regulieren 230. Es liegen jedoch nur wenige klinische Beweise für das Risiko der Behandlung von COVID-19-Patienten mit ARB vor oder ACE-Hemmer. Weitere Untersuchungen sind erforderlich, um herauszufinden, ob diese Medikamente den Viruseintritt hemmen oder unterstützen.
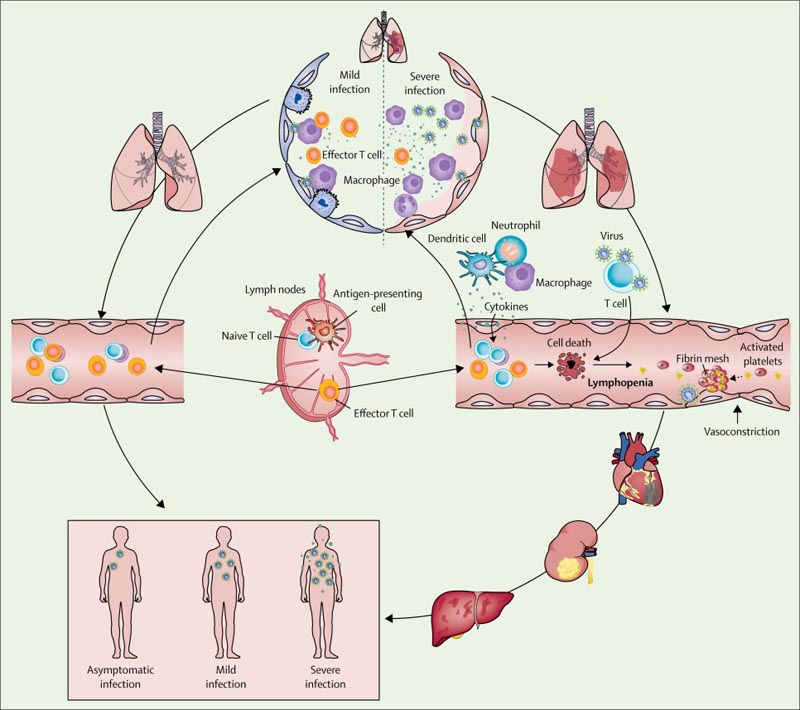 Abbildung: Auftreten und Entwicklung einer schweren Virussepsis mit akutem respiratorischem Syndrom und Coronavirus 2
Abbildung: Auftreten und Entwicklung einer schweren Virussepsis mit akutem respiratorischem Syndrom und Coronavirus 2
Schlussfolgerungen
Basierend auf Beobachtungen von Patienten mit COVID-19 stellten wir die Hypothese auf, dass in leichten Fällen residente Makrophagen, die pulmonale Entzündungsreaktionen auslösen, das Virus nach einer SARS-CoV-2-Infektion eindämmen konnten; Sowohl die angeborene als auch die adaptive Immunantwort wurden effizient aufgebaut, um die Virusreplikation einzudämmen, sodass sich der Patient schnell erholte.
In schweren oder kritischen Fällen von COVID-19 war jedoch die Integrität der Epithel-Endothel-Schranke (Luft-Blut-Schranke) stark gestört. Neben Epithelzellen kann SARS-CoV-2 auch Lungenkapillarendothelzellen angreifen, was zu einer großen Exsudatmenge von Plasmabestandteilen in die Alveolarhöhle führt.
Als Reaktion auf eine SARS-CoV-2-Infektion könnten Alveolarmakrophagen oder Epithelzellen verschiedene proinflammatorische Zytokine und Chemokine produzieren. Nach dieser Veränderung wirkten Monozyten und Neutrophile an der Infektionsstelle chemotaktisch, um diese Exsudate mit Viruspartikeln und infizierten Zellen zu beseitigen, was zu einer unkontrollierten Entzündung führte .
Bei diesem Prozess kann die adaptive Immunantwort aufgrund der erheblichen Reduktion und Funktionsstörung der Lymphozyten nicht effektiv eingeleitet werden. Eine unkontrollierte Virusinfektion führt zu einer erhöhten Makrophageninfiltration und einer weiteren Verschlechterung der Lungenschädigung.
In der Zwischenzeit führen ein direkter Angriff auf andere Organe durch disseminiertes SARS-CoV-2, eine durch einen systemischen Zytokinsturm verursachte Immunpathogenese und Mikrozirkulationsstörungen zusammen zu einer Virussepsis . Daher sind eine wirksame antivirale Therapie und Maßnahmen zur Modulation der angeborenen Immunantwort und zur Wiederherstellung der adaptiven Immunantwort unerlässlich, um den Teufelskreis zu durchbrechen und die Behandlungsergebnisse für die Patienten zu verbessern.
Seit dem Ausbruch von COVID-19 haben Ärzte ihr Bestes getan, um die Krankheit zu verstehen, und es wurde ein kurzes Porträt ihrer klinischen Merkmale erstellt. Es gibt jedoch noch offene Fragen zu den Mechanismen der Beobachtungen.
Mit Beweisen aus Autopsiestudien zu COVID-19 und wissenschaftlicher Grundlagenforschung zu SARS-CoV-2 und SARS-CoV haben wir nach mehreren Diskussionsrunden unter Forschern mehrere Hypothesen über die Pathogenese von SARS-CoV-2 aufgestellt. von Grundlagenwissenschaftlern, Pathologen und Klinikern, die an COVID-19 arbeiten.
Wir gehen davon aus, dass ein Prozess namens Virussepsis entscheidend für den Krankheitsmechanismus von COVID-19 ist. Obwohl sich diese Ideen später als unvollkommen oder sogar falsch herausstellen könnten, sind wir der Meinung, dass sie Fragen für zukünftige Forschung aufwerfen.
- Zukünftige wissenschaftliche Forschung ist erforderlich, um zu untersuchen, ob SARS-CoV-2 Gefäßendothelzellen direkt angreift, und um die Wirkung von SARS-CoV-2 auf die Gerinnung und Virusverbreitung zu untersuchen.
- Klinische Studien und Tierversuche sollten durchgeführt werden, um die Wirkung von ARB- und ACE-Hemmern auf den Ausgang einer SARS-CoV-2-Infektion in vivo zu bewerten.
- Es sollten Anstrengungen unternommen werden, um zu bestätigen, ob SARS-CoV-2 Lymphozyten direkt infiziert und wie es die adaptive Immunantwort beeinflusst.
- Auch die Kinetik der Zytokinreaktion während einer SARS-CoV-2-Infektion bedarf weiterer Untersuchungen.
- Die Wirksamkeit immunmodulatorischer Therapien sollte in randomisierten klinischen Studien evaluiert werden.















