Summary Since the outbreak of coronavirus disease 2019 (COVID-19), doctors have tried every effort to understand the disease, and a brief portrait of its clinical characteristics has been identified. In clinical practice, we noted that many severe or critically ill COVID-19 patients developed typical clinical manifestations of shock , including cold extremities and weak peripheral pulses, even in the absence of overt hypotension. Understanding the mechanism of viral sepsis in COVID-19 is the guarantee to explore better clinical care for these patients. With evidence collected from autopsy studies on COVID-19 and basic scientific research on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and SARS-CoV, we have put forward several hypotheses on the pathogenesis of SARS-CoV. -2 after multiple rounds of discussion between basic science researchers, pathologists and clinicians working on COVID-19. We hypothesize that a process called viral sepsis is crucial to the disease mechanism of COVID-19. Although these ideas may prove imperfect or even incorrect later, we believe they can provide input and direction for basic research at this time. |
The outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which was first reported in Wuhan, China, in December 2019, has had a huge impact on China and the entire world. The disease caused by SARS-CoV-2 is called coronavirus disease 2019 (COVID-19). By March 19, 2020, the number of confirmed cases had increased to more than 200,000. Although most patients infected with SARS-CoV-2 had mild illness, approximately 5% of patients had lung injury severe or even multiorgan dysfunction , resulting in a 1·4% fatality rate1.
In practice, we noted that many severe or critically ill COVID-19 patients developed typical clinical manifestations of shock, including cold extremities and weak peripheral pulses, even in the absence of overt hypotension. Many of these patients showed severe metabolic acidosis , indicating possible microcirculation dysfunction. Additionally, some patients had liver2 and kidney failure in addition to severe lung injury.
These patients met the diagnostic criteria for sepsis and septic shock according to the Sepsis-3 International Consensus 3 but SARS-CoV-2 infection appeared to be the only cause in most of them.1 Cultures of blood samples and of the lower respiratory tract were found to be negative for bacteria and fungi in 76% of patients with sepsis in a COVID-19 cohort.
Therefore, viral sepsis would be more accurate to describe the clinical manifestations of severe or critical COVID-19 patients.5 Understanding the mechanism of viral sepsis in COVID-19 is the guarantee to explore better clinical care for these patients.
Virus infection and pathogenesis of COVID-19 in organs
In biopsy or autopsy studies, lung pathology for both early6 and late phase7 COVID-19 patients showed diffuse alveolar damage with the formation of hyaline membranes, mononuclear cells and macrophages infiltrating air spaces, and a diffuse thickening of the alveolar wall. Viral particles were observed in type 2 bronchial alveolar epithelial cells by electron microscopy8, 9.
Furthermore, atrophy of the spleen, necrosis of hilar lymph nodes, focal hemorrhage in the kidney, enlarged liver with infiltration of inflammatory cells, edema, and scattered degeneration of neurons in the brain were present in some patients 8, 9.
Infectious SARS-CoV-2 virus particles have been isolated from respiratory samples,10 as well as fecal samples11 and urine (Zhao J, Guangzhou Medical University, personal communication) from patients with COVID-19, suggesting that dysfunction Multiple organ damage in severe COVID19 is at least partially caused by a direct attack by the virus. However, there are no reports on postmortem observations of the widespread dissemination of viral particles by autopsy at this time.
Whether SARS-CoV-2 can directly target organs other than the lung, especially those organs with high expression of angiotensin-converting enzyme 2 (ACE2) 12,13 and organs with L-SIGN(14) as possible alternative cellular receptors for SARS-CoV-2, it has to be investigated further. Furthermore, the question of how SARS-CoV-2 spreads to extra-pulmonary organs remains an enigma. Genomic variation of circulating SARS-CoV-2 has been observed, and the difference in virulence needs further investigation15.
Immune response to SARS-CoV-2 and viral sepsis
Proinflammatory cytokines and chemokines have been shown to include tumor necrosis factor (TNF) α, interleukin 1β (IL-1β), IL-6, granulocyte colony-stimulating factor, interferon gamma-induced protein 10, chemoattractant protein Monocyte and macrophage inflammatory proteins 1-α were significantly elevated in patients with COVID-19, 16, 17. As in severe influenza infection, cytokine storm could play an important role in the immunopathology of COVID-19. .
Previous studies revealed that lung epithelial cells, macrophages and dendritic cells all express cytokines to some extent during influenza infection through the activation of pattern recognition receptors (including Toll-like receptors TLR3, TLR7 and TLR8 ), the retinoic acid-inducible gene I and members of the NOD18-like receptor family.
However, little is known about the situation on COVID-19 at this time. It is crucial to identify the primary source of the cytokine storm in response to SARS-CoV-2 infection and the virological mechanisms behind the cytokine storm. It would also be relevant to elucidate the kinetics of cytokine activation during SARS-CoV-2 infection: When were the first cytokines released and what were they?
Furthermore, whether direct virus-induced tissue damage, systemic cytokine storm, or synergistic effects of both, contribute to the multi-organ dysfunction of severe COVID-19 patients remains to be addressed.
Additionally, it is worth monitoring whether blocking one of these proinflammatory mediators would affect the clinical outcome. Anti-IL-6R monoclonal antibody or corticosteroids have been proposed to alleviate the inflammatory response.
However, IL-6 could play an important role in initiating a preliminary response against virus infection by promoting neutrophil-mediated viral clearance, as one study revealed that deficiency of IL-6 or IL-6R led to the persistence of influenza infection and finally death in mice.19 And the use of corticosteroids remains controversial20, 21.
However, the dysregulated immune response also has an immune suppression stage after the proinflammatory phase. It is characterized by a sustained and substantial reduction in peripheral lymphocyte counts , mainly CD4 and CD8 T cells in patients with COVID-19, and is associated with a high risk of developing a secondary bacterial infection. This condition, known as lymphopenia , was also found in severe influenza and other respiratory viral infections.
The degree of lymphopenia has been shown to correlate with the severity of COVID-19.17
The mechanism underlying lymphopenia remains unknown. Previous studies have shown that SARS-like viral particles and SARS-CoV RNA were detected in T lymphocytes isolated from samples of peripheral blood, spleen, lymph nodes, and lymphoid tissue from various organs 22,23 suggesting that SARS-CoV could infect to T cells directly . The receptor binding domains of the spike proteins between SARS-CoV-2 and SARS-CoV show a high degree of consistency 24. 25 and SARS-CoV-2 RNA was also detected in blood samples16.
Therefore, it is reasonable to assume that, in addition to the activation-induced cell death induced by Fas and Fas ligand interaction, as well as the TNF-related apoptosis-inducing ligand axis, 26
SARS-CoV-2 could directly infect lymphocytes , particularly T cells, and initiate or promote lymphocyte cell death, eventually leading to lymphopenia and impaired antiviral responses. However, such a hypothesis needs to be investigated further.
It must also be identified what types of cell death are occurring in lymphocytes after SARS-CoV-2 infection. Furthermore, it is intriguing that lymphocytes lack ACE2 expression, suggesting an alternative mechanism by which SARS-CoV-2 engages T lymphocytes.13 Whether or not alveolar macrophages can phagocytose viral particles and then transfer them to lymphocytes is an open question in the field.
COVID-19 and abnormal coagulation
Studies have revealed that 71.4% of COVID-19 non-survivors reached the open disseminated intravascular coagulation grade (≥5 points according to the International Society on Thrombosis and Haemostasis criteria) 27 and showed abnormal coagulation results during the stages. later of the disease; Particularly increased concentrations of D-dimer and other fibrin degradation products were significantly associated with a poor prognosis4.
However, the specific mechanisms for coagulopathy have not yet been identified. Whether SARS-CoV-2 is able to directly attack vascular endothelial cells that express high levels of ACE2,13
and then lead to abnormal coagulation and sepsis remains to be explored.
Meanwhile, ACE2 is also an important blood pressure regulator . High expression of ACE2 in the circulatory system after SARS-CoV-2 infection could partially contribute to septic hypotension .
Questions have been raised about the use of angiotensin II receptor blocker (ARB) and ACE inhibitor therapy for patients with COVID-19 high blood pressure.
Some researchers suggested that ACE inhibitors could benefit these patients by reducing lung inflammation 29 although others argued that ACE inhibitors could improve viral entry by regulating ACE levels 230. However, there has been little clinical evidence on the risk of treating COVID-19 patients with ARB or ACE inhibitors. More research is needed to explore whether these medications inhibit or assist viral entry.
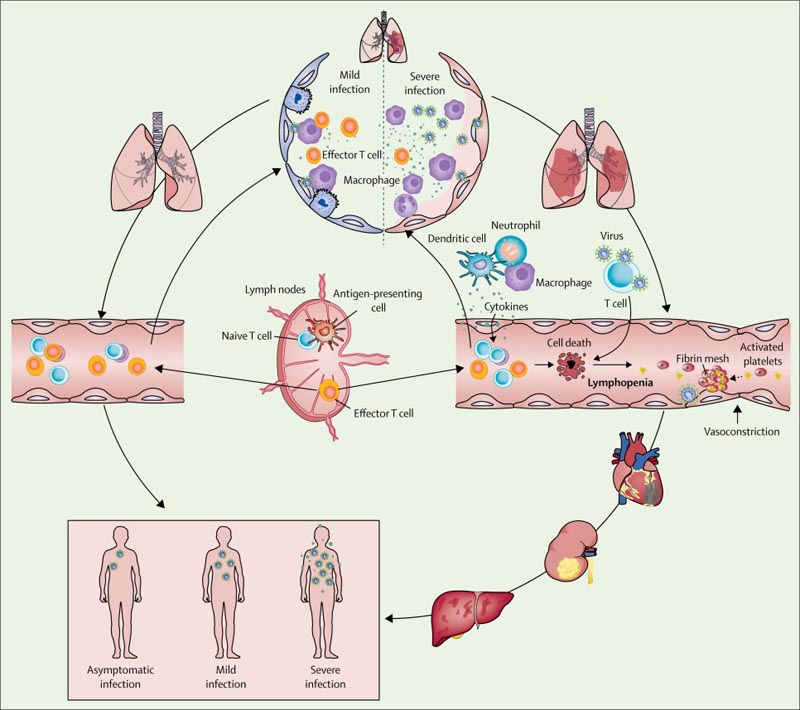 Figure Occurrence and evolution of severe acute respiratory syndrome coronavirus 2 viral sepsis
Figure Occurrence and evolution of severe acute respiratory syndrome coronavirus 2 viral sepsis
Conclusions
Based on observations of patients with COVID-19, we hypothesized that, in mild cases , resident macrophages that initiate pulmonary inflammatory responses were able to contain the virus after SARS-CoV-2 infection; Both innate and adaptive immune responses were efficiently established to curb viral replication so that the patient recovered quickly.
However, in severe or critical cases of COVID-19, the integrity of the epithelial-endothelial (air-blood) barrier was severely disrupted. In addition to epithelial cells, SARS-CoV-2 can also attack pulmonary capillary endothelial cells, leading to a large amount of exudate of plasma components into the alveolar cavity.
In response to SARS-CoV-2 infection, alveolar macrophages or epithelial cells could produce various proinflammatory cytokines and chemokines. Following this change, monocytes and neutrophils were chemotactic at the site of infection to eliminate these exudates with virus particles and infected cells, resulting in uncontrolled inflammation .
In this process, due to the substantial reduction and dysfunction of lymphocytes, the adaptive immune response cannot be initiated effectively. Uncontrolled viral infection leads to increased macrophage infiltration and further worsening of lung injury.
Meanwhile, direct attack on other organs by disseminated SARS-CoV-2, immune pathogenesis caused by systemic cytokine storm and microcirculation dysfunctions together lead to viral sepsis . Therefore, effective antiviral therapy and measures to modulate the innate immune response and restore the adaptive immune response are essential to break the vicious cycle and improve patient outcomes.
Since the outbreak of COVID-19, doctors have done their best to understand the disease, and a brief portrait of its clinical features has been identified. However, there are still open questions about the mechanisms of the observations.
With evidence collected from autopsy studies on COVID-19 and basic scientific research on SARS-CoV-2 and SARS-CoV, we have put forward several hypotheses about the pathogenesis of SARS-CoV-2 after multiple rounds of discussion among researchers. of basic sciences, pathologists, and clinicians working on COVID-19.
We hypothesize that a process called viral sepsis is crucial to the disease mechanism of COVID-19. Although these ideas could be proven imperfect or even incorrect later, we believe they raise questions for future research.
- Future scientific research is needed to explore whether SARS-CoV-2 directly attacks vascular endothelial cells and to examine the effect of SARS-CoV-2 on clotting and virus dissemination.
- Clinical trials and animal experiments should be performed to evaluate the effect of ARB and ACE inhibitors on the outcome of SARS-CoV-2 infection in vivo.
- Efforts should be made to confirm whether SARS-CoV-2 directly infects lymphocytes and how it influences the adaptive immune response.
- The kinetics of the cytokine response during SARS-CoV-2 infection also needs further investigation.
- The efficacy of immunomodulatory therapies should be evaluated in randomized clinical trials.















