
Contexte récapitulatif
L’efficacité de ces médicaments n’a pas encore été prouvée pour le traitement des maladies graves causées par le SRAS-CoV-2.
Méthodes
Nous avons mené un essai randomisé, contrôlé et ouvert incluant des patients adultes hospitalisés présentant une infection confirmée par le SRAS-CoV-2, responsable de la maladie respiratoire Covid-19, et une saturation en oxygène (Sao2) de 94 % ou moins lors de la respiration à température ambiante. . air ou un rapport de la pression partielle d’oxygène (Pao2) à la fraction d’oxygène inspiré (Fio2) inférieur à 300 mm Hg.
Les patients ont été répartis au hasard selon un rapport de 1:1 pour recevoir du lopinavir-ritonavir (400 mg et 100 mg, respectivement) deux fois par jour pendant 14 jours en plus des soins standard ou des soins standard seuls.
Le critère d’évaluation principal était le délai d’amélioration clinique, défini comme le temps écoulé entre la randomisation et une amélioration de deux points sur une échelle ordinale à sept catégories ou la sortie de l’hôpital, selon la première éventualité.
Résultats
Au total, 199 patients présentant une infection par le SRAS-CoV-2 confirmée en laboratoire ont été randomisés ; 99 ont été affectés au groupe lopinavir-ritonavir et 100 au groupe soins standards.
Le traitement par lopinavir-ritonavir n’a pas été associé à une différence par rapport aux soins standard en termes de délai d’amélioration clinique (risque relatif d’amélioration clinique, 1,24 ; intervalle de confiance [IC] à 95 %, 0,90 à 1,72).
La mortalité à 28 jours était similaire dans le groupe lopinavir-ritonavir et dans le groupe soins standards (19,2 % contre 25,0 % ; différence, −5,8 points de pourcentage ; IC à 95 %, −17,3 à 5,7) .
Les pourcentages de patients présentant un ARN viral détectable à différents moments étaient similaires.
Dans une analyse modifiée en intention de traiter, l’association lopinavir-ritonavir a entraîné une amélioration clinique médiane plus courte d’un jour que celle observée avec les soins standard (rapport de risque, 1,39 ; IC à 95 %, 1,00 à 1,91).
Les événements indésirables gastro-intestinaux étaient plus fréquents dans le groupe lopinavir-ritonavir, mais les événements indésirables graves étaient plus fréquents dans le groupe soins standards.
Le traitement par lopinavir-ritonavir a été arrêté prématurément chez 13 patients (13,8 %) en raison d’événements indésirables.
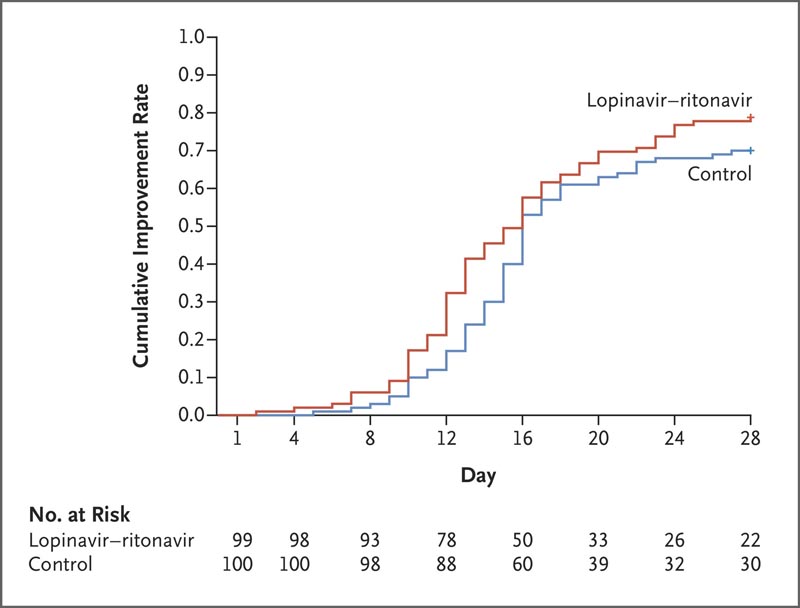
Délai d’amélioration clinique dans la population en intention de traiter.
Discussion
Cet essai randomisé a révélé que le traitement par lopinavir-ritonavir ajouté aux soins de soutien standard n’était pas associé à une amélioration clinique ou à une mortalité chez les patients gravement malades atteints de Covid-19 différente de celle associée aux soins standard seuls.
Cependant, dans l’analyse modifiée en intention de traiter, qui excluait trois patients décédés prématurément, la différence entre les groupes dans le délai médian d’amélioration clinique (médiane, 15 jours contre 16 jours) était significative, bien que modeste.
Il convient de noter que la mortalité globale dans cet essai (22,1 %) était nettement supérieure à la mortalité de 11 % à 14,5 % rapportée dans les premières études descriptives de patients hospitalisés atteints de Covid-19,1,2, ce qui indique que nous incluons une population gravement malade. . Notre population de patients était hétérogène en ce qui concerne la durée et la gravité de la maladie au moment de l’inscription ;
Une récupération clinique accélérée (16,0 jours contre 17,0 jours) et une réduction de la mortalité (19,0 % contre 27,1 %) ont été observées dans un sous-groupe post hoc de personnes traitées dans les 12 jours suivant l’apparition des symptômes, mais pas chez celles traitées plus tard. La question de savoir si un prétraitement par lopinavir-ritonavir dans le Covid-19 pourrait avoir un bénéfice clinique est importante et nécessite des études plus approfondies.
Cette découverte concorde avec les études montrant que les patients atteints de pneumonie virale du SRAS-CoV-2 présentent une progression au cours de la deuxième semaine de maladie et avec les effets du délai de traitement observés dans les études antivirales précédentes sur le SRAS-COV2 et la grippe grave. . De plus, nous avons constaté que le nombre de patients recevant du lopinavir-ritonavir ayant présenté des complications graves (lésion rénale aiguë et infections secondaires) ou ayant nécessité une ventilation mécanique invasive ou non invasive en raison d’une insuffisance respiratoire était inférieur à celui de ceux n’ayant pas reçu de traitement.
Ces observations sont génératrices d’hypothèses et nécessitent des études complémentaires pour déterminer si un traitement par lopinavir-ritonavir administré à un certain stade de la maladie peut réduire certaines complications du Covid-19.
Nous n’avons pas constaté que l’ajout d’un traitement par lopinavir-ritonavir réduisait les charges d’ARN viral ou la durée de détectabilité de l’ARN viral par rapport aux soins de soutien standard seuls. L’ARN du SRAS-CoV-2 était encore détecté chez 40,7 % des patients du groupe lopinavir-ritonavir à la fin de l’essai (jour 28). Un rapport récent a montré que la durée médiane d’excrétion du virus dans le cas du Covid-19 était de 20 jours chez les patients gravement malades et pouvait aller jusqu’à 37 jours.
Ni cette étude ni la présente étude n’ont trouvé de preuve que le lopinavir-ritonavir exerçait un effet antiviral significatif. Les raisons de l’absence apparente d’effet antiviral sont incertaines, mais les méthodes d’échantillonnage utilisées dans le présent essai n’étaient probablement pas optimales. Les échantillons n’ont été prélevés que par intermittence (jours 1, 5, 10, 14, 21 et 28), et des prélèvements plus fréquents au cours des 5 premiers jours auraient pu permettre une caractérisation plus détaillée de la cinétique de la charge virale dans les deux groupes pendant cette période critique. .
De plus, des études antérieures ont montré que les échantillons prélevés sur un écouvillon de gorge présentaient une charge virale inférieure à celle des échantillons nasopharyngés et, plus important encore, nous n’avons pas pu prélever les sécrétions des voies respiratoires inférieures.
Il convient de noter que selon le type de cellule utilisé, les concentrations efficaces à 50 % (CE50) du lopinavir in vitro pour le SRAS-CoV se situaient entre 4,0 et 10,7 μg par millilitre, 5,6,8, bien que d’autres études aient rapporté que le lopinavir était inactif26. ou des concentrations plus élevées (25 μg par millilitre) étaient nécessaires pour l’inhibition.
Pour le MERS-CoV, les valeurs EC50 variaient de 5 à environ 7 μg par millilitre. Les concentrations sériques maximales moyennes (9,6 μg par millilitre) et minimales (5,5 μg par millilitre) du lopinavir chez les adultes seuls se rapprochent de ces concentrations. On peut se demander si la valeur EC50 constitue un seuil approprié et si les concentrations de lopinavir non lié dans le plasma humain sont suffisantes pour l’inhibition du SRAS-CoV-2.
Près de 14 % des personnes ayant reçu du lopinavir-ritonavir n’ont pas pu terminer le traitement complet de 14 jours. Cela était principalement dû à des événements indésirables gastro-intestinaux , notamment l’anorexie, des nausées, des malaises abdominaux ou de la diarrhée, ainsi qu’à deux événements indésirables graves, tous deux une gastrite aiguë. Deux receveurs présentaient des éruptions cutanées spontanément résolutives.
De tels effets secondaires, notamment les risques de lésions hépatiques, de pancréatite, d’éruptions cutanées plus graves et d’allongement de l’intervalle QT, ainsi que le potentiel d’interactions médicamenteuses multiples dues à l’inhibition du CYP3A, sont bien documentés avec cette association médicamenteuse. Le profil d’effets secondaires observé dans l’essai actuel soulève des inquiétudes quant à l’utilisation de schémas posologiques de lopinavir-ritonavir plus élevés ou plus longs dans le but d’améliorer les résultats.
Conclusions
|
(Financé par les grands projets nationaux scientifiques et technologiques sur la création et le développement de nouveaux médicaments et autres ; numéro d’enregistrement d’essai clinique chinois, ChiCTR2000029308. Ouvre dans un nouvel onglet.)















