Utilizziamo i cookie per assicurarti la migliore esperienza di navigazione sul nostro sito web. Utilizzando il nostro sito, dichiari di aver letto e compreso la nostra Informativa sui cookie e Informativa sulla privacy
Il software identifica il potenziale di coinfezione con due varianti di coronavirus
Lo strumento VICOS aiuta a comprendere le coinfezioni con i ceppi esistenti.
I ricercatori argentini, che fanno parte del consorzio pubblico Proyecto País, hanno sviluppato un algoritmo che consente loro di identificare se una persona ha contratto due tipi di coronavirus SARS-CoV-2.
Lo strumento si chiama VICOS (Viral COinfection Surveillance) ed è stato utilizzato per rilevare coinfezioni SARS-CoV-2 nei dati di 1.097 genomi completi raccolti tra marzo 2020 e agosto 2021 in Argentina.
Secondo il lavoro, pubblicato sulla rivista Virus Research, sono stati rilevati 23 casi (2%) di diverse coinfezioni SARS-CoV-2. Se si analizzano i risultati, tre di questi casi erano dovuti a due virus dello stesso lignaggio, due erano dovuti a virus di diversi lignaggi genetici, 13 erano compatibili sia con la coinfezione che con l’evoluzione intra-ospite e 5 casi erano probabilmente il prodotto della contaminazione del laboratorio.
Dal 2020, i ricercatori del Proyecto País – un consorzio del Ministero della Scienza, Tecnologia e Innovazione della Nazione – hanno iniziato a sequenziare i genomi del coronavirus da campioni di pazienti affetti da Covid-19.
La nuova pipeline informatica VICOS ha rivelato il 2% delle coinfezioni da SARS-CoV-2 durante la prima ondata di COVID-19 in Argentina.
L’output di VICOS insieme a metadati aggiuntivi aiuta a distinguere tra 2 co-infezione SARS-CoV-2, evoluzione all’interno dell’ospite o contaminazione da sequenziamento.
Le coinfezioni SARS-CoV-2 interlineage e intralineage dovrebbero essere monitorate come forza trainante della ricombinazione e dell’evoluzione.
La coinfezione con due virus SARS-CoV-2 rimane un fenomeno molto poco studiato. Sebbene i metodi di sequenziamento di prossima generazione siano molto sensibili nel rilevare popolazioni virali eterogenee in un campione, non esiste un metodo standardizzato per la loro caratterizzazione, quindi la loro importanza clinica ed epidemiologica è sconosciuta.
VICOS (Viral COinfection Surveillance), un nuovo algoritmo di filtraggio bioinformatico e analisi statistica, è stato sviluppato per identificare campioni sospettati di essere popolazioni miste di SARS-CoV-2 da un ampio set di dati nel quadro della sorveglianza genomica comunitaria. VICOS è stato utilizzato per rilevare coinfezioni da SARS-CoV-2 in una serie di dati record di 1097 genomi interi raccolti tra marzo 2020 e agosto 2021 in Argentina.
Sono stati rilevati 23 casi (2%) di coinfezioni da SARS-CoV-2. Uno studio dettagliato dei risultati del VICOS insieme ad un’ulteriore analisi filogenetica ha rivelato 3 casi di coinfezioni da parte di due virus della stessa linea genetica, 2 casi di virus di diverse linee genetiche, 13 erano compatibili sia con la coinfezione che con l’evoluzione intra-ospite e 5 casi erano probabilmente prodotto di contaminazione da laboratorio.
La diversità virale all’interno del campione fornisce informazioni importanti per comprendere le dinamiche di trasmissione di SARS-CoV-2. Gli strumenti bioinformatici avanzati, come VICOS, sono una risorsa necessaria per aiutare a rivelare la pluralità nascosta di SARS-CoV-2.
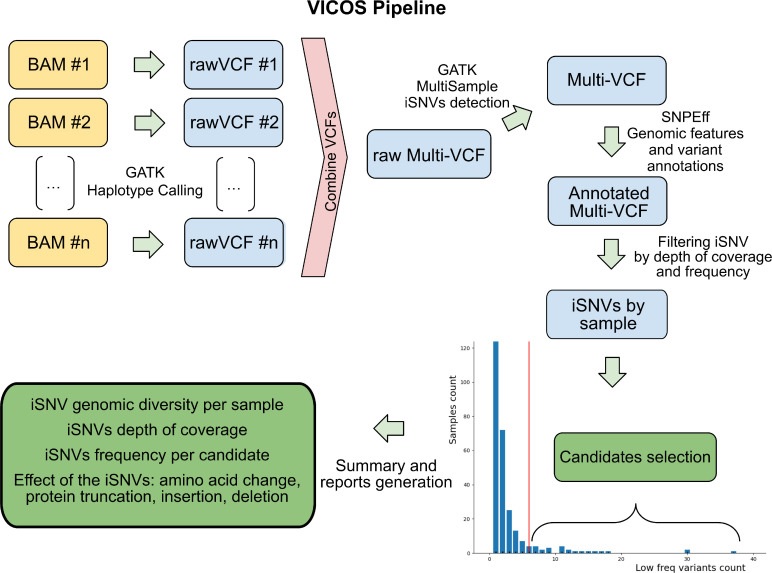
Algoritmo funzionante utilizzato da VICOS per identificare e riportare il numero, la frequenza e la posizione degli iSNV nei genomi SARS-CoV-2 ottenuti con la tecnologia Illumina. La pipeline richiede un set di file BAM come input.