Nous utilisons des cookies pour vous garantir la meilleure expérience de navigation sur notre site Internet. En utilisant notre site, vous reconnaissez avoir lu et compris notre Politique en matière de cookies
Un logiciel identifie un potentiel de co-infection avec deux variantes du coronavirus
L'outil VICOS aide à comprendre les co-infections avec les souches existantes.
Des chercheurs argentins, qui font partie du consortium public Proyecto País, ont développé un algorithme qui leur permet d’identifier si une personne a contracté deux types de coronavirus SARS-CoV-2.
L’outil s’appelle VICOS (Viral COinfection Surveillance) et a été utilisé pour détecter les co-infections par le SRAS-CoV-2 dans les données de 1 097 génomes complets collectés entre mars 2020 et août 2021 en Argentine.
Selon les travaux, publiés dans la revue Virus Research, 23 cas (2 %) de différentes co-infections par le SRAS-CoV-2 ont été détectés. Si l’on décompose les résultats, trois de ces cas étaient dus à deux virus de la même lignée, deux étaient dus à des virus de lignées génétiques différentes, 13 étaient compatibles à la fois avec une co-infection et une évolution intra-hôte, et 5 cas étaient probablement le produit de contamination du laboratoire.
Depuis 2020, des chercheurs du Proyecto País – un consortium du ministère de la Science, de la Technologie et de l’Innovation de la Nation – ont commencé à séquencer les génomes des coronavirus à partir d’échantillons de patients atteints de Covid-19.
Le nouveau pipeline informatique VICOS a révélé 2 % de co-infections par le SRAS-CoV-2 lors de la première vague de COVID-19 en Argentine.
La sortie VICOS ainsi que des métadonnées supplémentaires aident à différencier 2 co-infections par le SRAS-CoV-2, l’évolution au sein de l’hôte ou la contamination par séquençage.
Les co-infections interlignées et intralignées par le SRAS-CoV-2 doivent être surveillées en tant que force motrice de la recombinaison et de l’évolution.
La co-infection par deux virus SARS-CoV-2 reste un phénomène très peu étudié. Bien que les méthodes de séquençage de nouvelle génération soient très sensibles pour détecter des populations virales hétérogènes dans un échantillon, il n’existe aucune méthode standardisée pour leur caractérisation, de sorte que leur importance clinique et épidémiologique est inconnue.
VICOS (Viral COinfection Surveillance), un nouvel algorithme de filtrage bioinformatique et d’analyse statistique, a été développé pour identifier des échantillons suspectés d’être des populations mixtes de SRAS-CoV-2 à partir d’un vaste ensemble de données dans le cadre de la surveillance génomique communautaire. VICOS a été utilisé pour détecter les co-infections par le SRAS-CoV-2 dans un ensemble de données d’enregistrement provenant de 1 097 génomes entiers collectés entre mars 2020 et août 2021 en Argentine.
23 cas (2 %) de co-infections par le SRAS-CoV-2 ont été détectés. L’étude détaillée des résultats de VICOS ainsi qu’une analyse phylogénétique complémentaire ont révélé 3 cas de co-infections par deux virus de la même lignée, 2 cas par des virus de lignées génétiques différentes, 13 étaient compatibles à la fois avec une co-infection et une évolution intra-hôte, et 5 cas étaient probablement produit de contamination en laboratoire.
La diversité virale au sein de l’échantillon fournit des informations importantes pour comprendre la dynamique de transmission du SRAS-CoV-2. Les outils bioinformatiques avancés, tels que VICOS, sont une ressource nécessaire pour aider à révéler la pluralité cachée du SRAS-CoV-2.
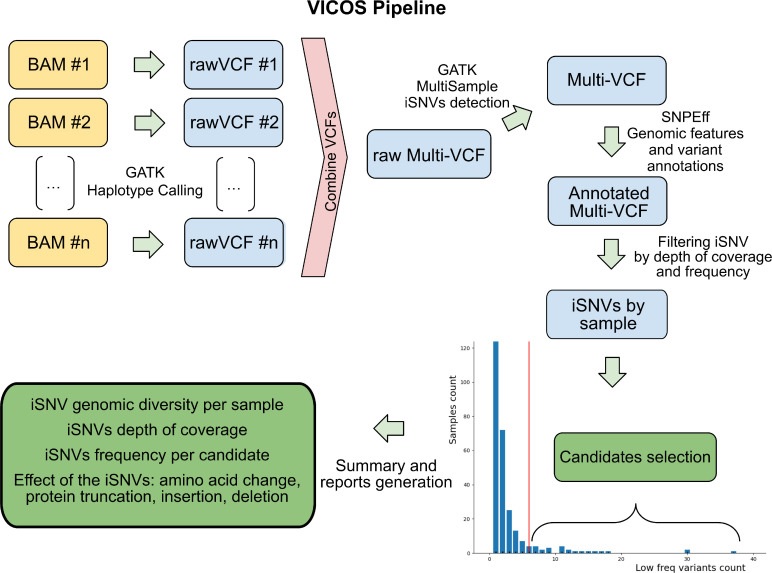
Algorithme de travail utilisé par VICOS pour identifier et signaler le nombre, la fréquence et la position des iSNV dans les génomes du SRAS-CoV-2 obtenus avec la technologie Illumina. Le pipeline nécessite un ensemble de fichiers BAM en entrée.