
ISTITUTO DI RICERCA SCRIPPS
L’analisi di Scripps Research dei dati pubblici sulla sequenza del genoma per SARS-CoV-2 e virus correlati non ha trovato prove che il virus sia stato prodotto in un laboratorio o sia stato ingegnerizzato
Secondo i risultati, il nuovo coronavirus SARS-CoV-2 emerso nella città di Wuhan, in Cina, l’anno scorso e che da allora ha causato un’epidemia di COVID-19 su larga scala diffondendosi in più di 70 paesi, è il prodotto di un’evoluzione naturale . pubblicato sulla rivista Nature Medicine .
L’analisi dei dati pubblici sulla sequenza del genoma della SARS-CoV-2 e dei virus correlati non ha trovato prove che il virus sia stato prodotto in laboratorio o altrimenti ingegnerizzato.
"Confrontando i dati disponibili sulla sequenza del genoma per i ceppi di coronavirus noti, possiamo determinare con certezza che SARS-CoV-2 ha avuto origine attraverso processi naturali", ha affermato Kristian Andersen, PhD, professore associato di immunologia e microbiologia presso Scripps Research e autore corrispondente dell’articolo. .
Oltre ad Andersen, gli autori dell’articolo "L’origine prossima della SARS-CoV-2" includono Robert F. Garry della Tulane University; Edward Holmes, Università di Sydney; Andrew Rambaut, dell’Università di Edimburgo; W. Ian Lipkin, Columbia University.
I coronavirus sono una vasta famiglia di virus che possono causare malattie di gravità molto variabile. La prima malattia grave causata da un coronavirus è emersa con l’epidemia di sindrome respiratoria acuta grave (SARS) del 2003 in Cina. Una seconda epidemia grave è iniziata nel 2012 in Arabia Saudita con la sindrome respiratoria del Medio Oriente (MERS).
Il 31 dicembre dello scorso anno, le autorità cinesi hanno allertato l’Organizzazione mondiale della sanità dell’epidemia di un nuovo ceppo di coronavirus che causa una malattia grave, in seguito denominata SARS-CoV-2. Al 20 febbraio 2020, sono stati documentati quasi 167.500 casi di COVID-19, anche se molti casi più lievi probabilmente non sono stati diagnosticati. Il virus ha ucciso più di 6.600 persone.
Poco dopo l’inizio dell’epidemia, gli scienziati cinesi hanno sequenziato il genoma della SARS-CoV-2 e hanno reso i dati disponibili ai ricercatori di tutto il mondo.
I dati risultanti dalla sequenza genomica hanno dimostrato che le autorità cinesi hanno rilevato rapidamente l’epidemia e che il numero di casi di COVID-19 è aumentato a causa della trasmissione da uomo a uomo dopo una singola introduzione nella popolazione umana. Andersen e i suoi collaboratori presso diversi altri istituti di ricerca hanno utilizzato questi dati di sequenziamento per esplorare le origini e l’evoluzione della SARS-CoV-2 concentrandosi su diverse caratteristiche rivelatrici del virus.
Gli scienziati hanno analizzato il modello genetico delle proteine spike , l’armatura esterna del virus che utilizza per intrappolare e penetrare nelle pareti esterne delle cellule umane e animali. Più specificamente, si sono concentrati su due importanti caratteristiche della proteina spike: il dominio di legame del recettore (RBD), un tipo di rampino che si attacca alle cellule ospiti, e il sito di clivaggio, un apriscatole molecolare che consente al virus di aprirsi. ed entra nelle cellule ospiti.
Prova dell’evoluzione naturale
Gli scienziati hanno scoperto che la porzione RBD delle proteine spike del SARS-CoV-2 si era evoluta per colpire efficacemente una caratteristica molecolare all’esterno delle cellule umane chiamata ACE2 , un recettore coinvolto nella regolazione della pressione sanguigna. La proteina spike del SARS-CoV-2 è stata così efficace nel legarsi alle cellule umane, infatti, che gli scienziati hanno concluso che fosse il risultato della selezione naturale e non il prodotto dell’ingegneria genetica .
Questa prova dell’evoluzione naturale è stata supportata dai dati sulla spina dorsale della SARS-CoV-2: la sua struttura molecolare complessiva. Se qualcuno avesse cercato di progettare un nuovo coronavirus come agente patogeno, lo avrebbe costruito partendo dalla spina dorsale di un virus noto per causare malattie. Ma gli scienziati hanno scoperto che la struttura portante del SARS-CoV-2 differiva sostanzialmente da quella dei coronavirus già noti e somigliava per lo più a virus correlati presenti nei pipistrelli e nei pangolini.
"Queste due caratteristiche del virus, le mutazioni nella porzione RBD della proteina spike e la sua distinta spina dorsale, escludono la manipolazione di laboratorio come possibile origine della SARS-CoV-2", ha affermato Andersen.
Josie Golding, PhD, leader dell’epidemia presso il Wellcome Trust con sede nel Regno Unito, ha affermato che le scoperte di Andersen e dei suoi colleghi sono "di fondamentale importanza nel fornire una visione basata sull’evidenza delle voci che circolano sulle origini". del virus (SARS-CoV)-2) che causa il COVID-19."
"Concludono che il virus è un prodotto dell’evoluzione naturale", aggiunge Goulding, "ponendo fine a qualsiasi speculazione sull’ingegneria genetica deliberata".
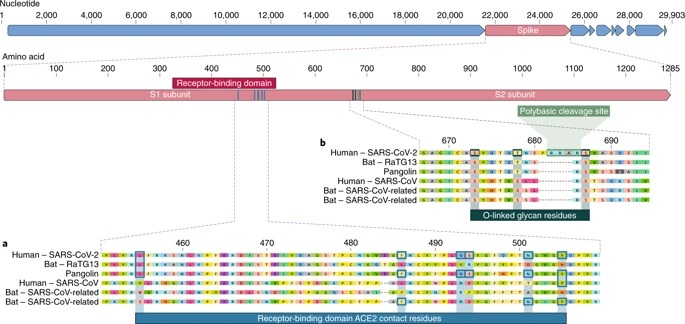
a, Mutazioni nei residui di contatto della proteina spike SARS-CoV-2. La proteina del picco SARS-CoV-2 (barra rossa in alto) era allineata con i coronavirus simili a SARS-CoV più strettamente correlati e con il SARS-CoV stesso. I residui chiave sulla proteina spike che contattano il recettore ACE2 sono contrassegnati con riquadri blu sia nella SARS-CoV-2 che nei virus correlati, incluso la SARS-CoV (ceppo Urbani). b, Acquisizione del sito di scissione polibasico e dei glicani legati all’O. Sia il sito di scissione polibasico che i tre glicani adiacenti legati all’O previsti sono unici per SARS-CoV-2 e non erano stati precedentemente osservati nei betacoronavirus della linea B. Le sequenze mostrate provengono da NCBI GenBank, codici di accesso MN908947, MN996532, AY278741, KY417146 e MK211376. Le sequenze del coronavirus del pangolino sono un consenso generato da SRR10168377 e SRR10168378 (NCBI BioProject PRJNA573298) 29,30.
Possibili origini del virus
Sulla base della loro analisi di sequenziamento genomico, Andersen e i suoi collaboratori hanno concluso che le origini più probabili della SARS-CoV-2 seguivano uno dei due possibili scenari.
In uno scenario, il virus si è evoluto fino al suo attuale stato patogeno attraverso la selezione naturale in un ospite non umano e poi è passato all’uomo . È così che sono emerse precedenti epidemie di coronavirus, con gli esseri umani che hanno contratto il virus dopo l’esposizione diretta a zibetti (SARS) e cammelli (MERS). I ricercatori hanno proposto che i pipistrelli siano il serbatoio più probabile della SARS-CoV-2, poiché è molto simile al coronavirus dei pipistrelli. Tuttavia, non ci sono casi documentati di trasmissione diretta pipistrello-uomo, suggerendo che probabilmente fosse coinvolto un ospite intermedio tra pipistrelli e umani.
In questo scenario, le due caratteristiche distintive della proteina spike del SARS-CoV-2, la porzione RBD che si lega alle cellule e il sito di clivaggio che apre il virus, si sarebbero evolute fino al loro stato attuale prima di entrare nell’uomo. . In questo caso, l’attuale epidemia sarebbe probabilmente scoppiata rapidamente non appena gli esseri umani fossero stati infettati, poiché il virus avrebbe già sviluppato le caratteristiche che lo rendono patogeno e capace di diffondersi tra le persone.
Nell’altro scenario proposto, una versione non patogena del virus è passata da un ospite animale all’uomo e poi si è evoluta fino al suo attuale stato patogeno all’interno della popolazione umana. Ad esempio, alcuni coronavirus dei pangolini, mammiferi simili agli armadilli presenti in Asia e Africa, hanno una struttura RBD molto simile a quella del SARS-CoV-2. Un coronavirus proveniente da un pangolino potrebbe essere stato trasmesso a un essere umano, direttamente o attraverso un ospite intermedio, come zibetti o furetti.
Quindi l’altra caratteristica proteina spike della SARS-CoV-2, il sito di scissione, potrebbe essersi evoluta all’interno di un ospite umano, possibilmente attraverso una circolazione limitata e non rilevata nella popolazione umana prima dell’inizio dell’epidemia.
I ricercatori hanno scoperto che il sito di clivaggio del SARS-CoV-2 appare simile ai siti di clivaggio dei ceppi di influenza aviaria che hanno dimostrato di diffondersi facilmente tra le persone. SARS-CoV-2 avrebbe potuto sviluppare un sito di scissione così virulento nelle cellule umane e presto dare inizio all’attuale epidemia, poiché il coronavirus sarebbe forse diventato molto più capace di diffondersi tra le persone.
Il coautore dello studio Andrew Rambaut ha avvertito che è difficile, se non impossibile, sapere a questo punto quale scenario sia più probabile. Se il SARS-CoV-2 entrasse nell’uomo nella sua attuale forma patogena da una fonte animale, la probabilità di future epidemie aumenterebbe, poiché il ceppo virale che causa la malattia potrebbe ancora circolare nella popolazione animale e potrebbe riemergere. saltare gli umani . Sono minori le probabilità che un coronavirus non patogeno entri nella popolazione umana e quindi sviluppi proprietà simili al SARS-CoV-2.















