
SCRIPPS FORSCHUNGSINSTITUT
Die Analyse öffentlicher Genomsequenzdaten für SARS-CoV-2 und verwandte Viren durch Scripps Research ergab keine Hinweise darauf, dass das Virus in einem Labor hergestellt oder manipuliert wurde
Den Ergebnissen zufolge ist das neue Coronavirus SARS-CoV-2, das letztes Jahr in der Stadt Wuhan, China, aufgetreten ist und seitdem eine großflächige COVID-19-Epidemie verursacht und sich in mehr als 70 Ländern ausgebreitet hat, ein Produkt der natürlichen Evolution veröffentlicht in der Zeitschrift Nature Medicine .
Die Analyse öffentlicher Genomsequenzdaten für SARS-CoV-2 und verwandte Viren ergab keine Hinweise darauf, dass das Virus in einem Labor hergestellt oder auf andere Weise manipuliert wurde.
„Durch den Vergleich der verfügbaren Genomsequenzdaten bekannter Coronavirus-Stämme können wir eindeutig feststellen, dass SARS-CoV-2 durch natürliche Prozesse entstanden ist“, sagte Kristian Andersen, PhD, außerordentlicher Professor für Immunologie und Mikrobiologie bei Scripps Research und korrespondierender Autor des Papiers .
Zu den Autoren des Papiers „The proximate origin of SARS-CoV-2“ gehören neben Andersen auch Robert F. Garry von der Tulane University; Edward Holmes, Universität Sydney; Andrew Rambaut von der University of Edinburgh; W. Ian Lipkin, Columbia University.
Coronaviren sind eine große Familie von Viren, die Erkrankungen unterschiedlicher Schwere verursachen können. Die erste bekannte schwere Krankheit, die durch ein Coronavirus verursacht wurde, trat 2003 mit der SARS-Epidemie (Schweres Akutes Atemwegssyndrom) in China auf. Ein zweiter Ausbruch einer schweren Krankheit begann 2012 in Saudi-Arabien mit dem Middle East Respiratory Syndrome (MERS).
Am 31. Dezember letzten Jahres alarmierten chinesische Behörden die Weltgesundheitsorganisation über den Ausbruch eines neuen Coronavirus-Stammes, der schwere Krankheiten verursacht und später den Namen SARS-CoV-2 erhielt. Bis zum 20. Februar 2020 wurden fast 167.500 Fälle von COVID-19 dokumentiert, obwohl viele mildere Fälle wahrscheinlich nicht diagnostiziert wurden. Das Virus hat mehr als 6.600 Menschen getötet.
Kurz nach Beginn der Epidemie sequenzierten chinesische Wissenschaftler das Genom von SARS-CoV-2 und stellten die Daten Forschern auf der ganzen Welt zur Verfügung.
Die daraus resultierenden Genomsequenzdaten haben gezeigt, dass die chinesischen Behörden die Epidemie schnell erkannten und dass die Zahl der COVID-19-Fälle aufgrund der Übertragung von Mensch zu Mensch nach einmaliger Einschleppung in die menschliche Bevölkerung gestiegen ist. Andersen und ihre Mitarbeiter an mehreren anderen Forschungseinrichtungen nutzten diese Sequenzierungsdaten, um den Ursprung und die Entwicklung von SARS-CoV-2 zu erforschen, indem sie sich auf mehrere verräterische Merkmale des Virus konzentrierten.
Die Wissenschaftler analysierten die genetische Vorlage für Spike-Proteine , eine Panzerung an der Außenseite des Virus, mit der es die Außenwände menschlicher und tierischer Zellen einfängt und durchdringt. Genauer gesagt konzentrierten sie sich auf zwei wichtige Merkmale des Spike-Proteins: die Rezeptorbindungsdomäne (RBD), eine Art Enterhaken, der sich an Wirtszellen bindet, und die Spaltungsstelle, ein molekularer Dosenöffner, der es dem Virus ermöglicht, aufzubrechen und in Wirtszellen eindringen.
Beweis der natürlichen Evolution
Die Wissenschaftler entdeckten, dass sich der RBD-Anteil der SARS-CoV-2-Spike-Proteine so entwickelt hatte, dass er effektiv auf ein molekulares Merkmal an der Außenseite menschlicher Zellen namens ACE2 abzielt , einen Rezeptor, der an der Regulierung des Blutdrucks beteiligt ist. Das SARS-CoV-2-Spike-Protein war bei der Bindung an menschliche Zellen tatsächlich so effektiv, dass Wissenschaftler zu dem Schluss kamen, es sei das Ergebnis natürlicher Selektion und nicht das Produkt der Gentechnik .
Dieser Beweis einer natürlichen Evolution wurde durch Daten zum Rückgrat von SARS-CoV-2 gestützt: seiner gesamten molekularen Struktur. Wenn jemand ein neues Coronavirus als Krankheitserreger entwickeln wollte, hätte er es aus dem Rückgrat eines Virus aufgebaut, von dem bekannt ist, dass es Krankheiten verursacht. Wissenschaftler fanden jedoch heraus, dass sich das Rückgrat von SARS-CoV-2 erheblich von dem bereits bekannter Coronaviren unterschied und größtenteils verwandten Viren ähnelte, die in Fledermäusen und Schuppentieren vorkommen.
„Diese beiden Merkmale des Virus, Mutationen im RBD-Teil des Spike-Proteins und sein ausgeprägtes Rückgrat, schließen Labormanipulationen als möglichen Ursprung von SARS-CoV-2 aus“, sagte Andersen.
Josie Golding, PhD, Epidemieleiterin beim britischen Wellcome Trust, sagte, die Ergebnisse von Andersen und ihren Kollegen seien „von entscheidender Bedeutung, um eine evidenzbasierte Sicht auf die Gerüchte zu liefern, die über die Ursprünge im Umlauf waren .“ des Virus (SARS-CoV)-2), das COVID-19 verursacht.“
„Sie kommen zu dem Schluss, dass das Virus ein Produkt der natürlichen Evolution ist“, fügt Goulding hinzu, „und beendet damit alle Spekulationen über absichtliche Gentechnik.“
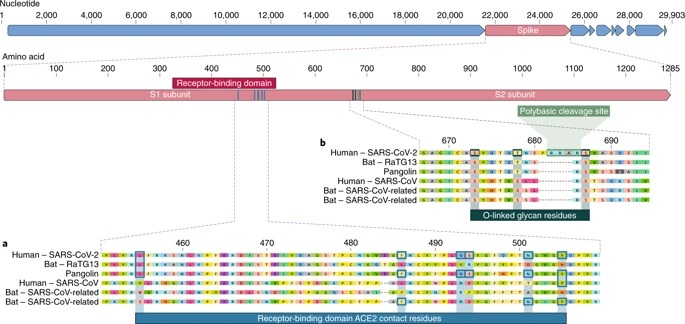
a, Mutationen in den Kontaktresten des SARS-CoV-2-Spike-Proteins. Das SARS-CoV-2-Spike-Protein (roter Balken oben) wurde gegen die enger verwandten SARS-CoV-ähnlichen Coronaviren und SARS-CoV selbst ausgerichtet. Wichtige Rückstände auf dem Spike-Protein, die mit dem ACE2-Rezeptor in Kontakt treten, sind sowohl bei SARS-CoV-2 als auch bei verwandten Viren, einschließlich SARS-CoV (Urbani-Stamm), mit blauen Kästchen markiert. b, Erwerb der polybasischen Spaltstelle und der O-verknüpften Glykane. Sowohl die polybasische Spaltstelle als auch die drei benachbarten vorhergesagten O-verknüpften Glykane kommen nur bei SARS-CoV-2 vor und wurden bisher bei Betacoronaviren der B-Linie nicht beobachtet. Die gezeigten Sequenzen stammen von NCBI GenBank, Zugangscodes MN908947, MN996532, AY278741, KY417146 und MK211376. Die Pangolin-Coronavirus-Sequenzen sind ein Konsens, der aus SRR10168377 und SRR10168378 (NCBI BioProject PRJNA573298) generiert wurde 29,30.
Mögliche Ursprünge des Virus
Basierend auf ihrer Genomsequenzierungsanalyse kamen Andersen und seine Mitarbeiter zu dem Schluss, dass der wahrscheinlichste Ursprung von SARS-CoV-2 einem von zwei möglichen Szenarien folgte.
In einem Szenario entwickelte sich das Virus durch natürliche Selektion in einem nichtmenschlichen Wirt zu seinem aktuellen pathogenen Zustand und sprang dann auf den Menschen über . So kam es zu früheren Coronavirus-Ausbrüchen, bei denen sich Menschen nach direktem Kontakt mit Zibetkatzen (SARS) und Kamelen (MERS) mit dem Virus infizierten. Die Forscher schlugen Fledermäuse als wahrscheinlichstes Reservoir für SARS-CoV-2 vor, da es einem Fledermaus-Coronavirus sehr ähnlich ist. Allerdings gibt es keine dokumentierten Fälle einer direkten Fledermaus-Mensch-Übertragung, was darauf hindeutet, dass es sich wahrscheinlich um einen Zwischenwirt zwischen Fledermäusen und Menschen handelte.
In diesem Szenario hätten sich die beiden Unterscheidungsmerkmale des SARS-CoV-2-Spike-Proteins, der RBD-Teil, der an Zellen bindet, und die Spaltstelle, die das Virus öffnet, zu ihrem aktuellen Zustand entwickelt, bevor sie in den Menschen gelangten. . In diesem Fall wäre die aktuelle Epidemie wahrscheinlich schnell ausgebrochen, sobald Menschen infiziert worden wären, da das Virus bereits die Eigenschaften entwickelt hätte, die es pathogen machen und sich zwischen Menschen ausbreiten könnten.
Im anderen vorgeschlagenen Szenario sprang eine nicht pathogene Version des Virus von einem tierischen Wirt auf den Menschen über und entwickelte sich dann innerhalb der menschlichen Bevölkerung zu ihrem aktuellen pathogenen Zustand. Beispielsweise haben einige Coronaviren von Schuppentieren, gürteltierähnlichen Säugetieren, die in Asien und Afrika vorkommen, eine RBD-Struktur, die der von SARS-CoV-2 sehr ähnlich ist. Ein von einem Schuppentier stammendes Coronavirus könnte entweder direkt oder über einen Zwischenwirt wie Zibetkatzen oder Frettchen auf einen Menschen übertragen worden sein.
Das andere charakteristische Spike-Protein von SARS-CoV-2, die Spaltungsstelle, könnte sich also in einem menschlichen Wirt entwickelt haben, möglicherweise durch eine begrenzte, unentdeckte Zirkulation in der menschlichen Bevölkerung vor Beginn der Epidemie.
Die Forscher fanden heraus, dass die Spaltstelle von SARS-CoV-2 den Spaltstellen von Vogelgrippestämmen ähnelt, die sich nachweislich leicht zwischen Menschen verbreiten. SARS-CoV-2 könnte eine solche virulente Spaltstelle in menschlichen Zellen entwickelt und bald die aktuelle Epidemie ausgelöst haben, da das Coronavirus möglicherweise viel besser in der Lage gewesen wäre, sich zwischen Menschen auszubreiten.
Der Co-Autor der Studie, Andrew Rambaut, warnte, dass es zum jetzigen Zeitpunkt schwierig, wenn nicht unmöglich sei, zu wissen, welches Szenario am wahrscheinlichsten sei. Wenn SARS-CoV-2 in seiner aktuellen pathogenen Form aus einer tierischen Quelle auf den Menschen gelangt, steigt die Wahrscheinlichkeit zukünftiger Ausbrüche, da der Virusstamm, der die Krankheit verursacht, möglicherweise immer noch in der Tierpopulation zirkuliert und erneut auftritt. springe Menschen . Die Wahrscheinlichkeit, dass ein nicht pathogenes Coronavirus in die menschliche Bevölkerung gelangt und dann ähnliche Eigenschaften wie SARS-CoV-2 entwickelt, ist geringer.















