
INSTITUT DE RECHERCHE SCRIPPS
L’analyse par Scripps Research des données publiques sur la séquence du génome du SRAS-CoV-2 et des virus apparentés n’a trouvé aucune preuve que le virus a été produit en laboratoire ou a été modifié.
Le nouveau coronavirus SARS-CoV-2, apparu l’année dernière dans la ville de Wuhan, en Chine, et qui a depuis provoqué une épidémie de COVID-19 à grande échelle et s’est propagé à plus de 70 pays, est un produit de l’ évolution naturelle , selon les résultats. publié dans la revue Nature Medicine .
L’analyse des données publiques sur la séquence du génome du SRAS-CoV-2 et des virus apparentés n’a trouvé aucune preuve que le virus ait été produit en laboratoire ou autrement modifié.
"En comparant les données disponibles sur la séquence du génome des souches connues de coronavirus, nous pouvons déterminer avec certitude que le SRAS-CoV-2 est né de processus naturels", a déclaré Kristian Andersen, PhD, professeur agrégé d’immunologie et de microbiologie à Scripps Research et auteur correspondant de l’article. .
Outre Andersen, les auteurs de l’article « L’origine immédiate du SRAS-CoV-2 » comprennent Robert F. Garry de l’Université de Tulane ; Edward Holmes, Université de Sydney ; Andrew Rambaut, de l’Université d’Édimbourg ; W. Ian Lipkin, Université de Columbia.
Les coronavirus constituent une grande famille de virus pouvant provoquer des maladies de gravité très variable. La première maladie grave connue causée par un coronavirus est apparue en 2003 avec l’épidémie du syndrome respiratoire aigu sévère (SRAS) en Chine. Une deuxième épidémie de maladie grave a débuté en 2012 en Arabie saoudite avec le syndrome respiratoire du Moyen-Orient (MERS).
Le 31 décembre de l’année dernière, les autorités chinoises ont alerté l’Organisation mondiale de la santé concernant l’apparition d’une nouvelle souche de coronavirus provoquant une maladie grave, qui a ensuite été baptisée SARS-CoV-2. Au 20 février 2020, près de 167 500 cas de COVID-19 avaient été recensés, même si de nombreux cas plus bénins n’ont probablement pas été diagnostiqués. Le virus a tué plus de 6 600 personnes.
Peu de temps après le début de l’épidémie, des scientifiques chinois ont séquencé le génome du SRAS-CoV-2 et mis les données à la disposition des chercheurs du monde entier.
Les données de séquence génomique obtenues ont montré que les autorités chinoises ont rapidement détecté l’épidémie et que le nombre de cas de COVID-19 a augmenté en raison de la transmission interhumaine après une seule introduction dans la population humaine. Andersen et ses collaborateurs de plusieurs autres instituts de recherche ont utilisé ces données de séquençage pour explorer les origines et l’évolution du SRAS-CoV-2 en se concentrant sur plusieurs caractéristiques révélatrices du virus.
Les scientifiques ont analysé le modèle génétique des protéines Spike , une armure située à l’extérieur du virus qu’il utilise pour piéger et pénétrer les parois externes des cellules humaines et animales. Plus précisément, ils se sont concentrés sur deux caractéristiques importantes de la protéine Spike : le domaine de liaison au récepteur (RBD), un type de grappin qui s’attache aux cellules hôtes, et le site de clivage, un ouvre-boîte moléculaire qui permet au virus de s’ouvrir. et pénètre dans les cellules hôtes.
Preuve de l’évolution naturelle
Les scientifiques ont découvert que la partie RBD des protéines de pointe du SRAS-CoV-2 avait évolué pour cibler efficacement une caractéristique moléculaire à l’extérieur des cellules humaines appelée ACE2 , un récepteur impliqué dans la régulation de la pression artérielle. En fait, la protéine Spike du SRAS-CoV-2 était si efficace pour se lier aux cellules humaines que les scientifiques ont conclu qu’elle était le résultat de la sélection naturelle et non du génie génétique .
Cette preuve d’évolution naturelle a été étayée par des données sur l’épine dorsale du SRAS-CoV-2 : sa structure moléculaire globale. Si quelqu’un cherchait à créer un nouveau coronavirus comme agent pathogène, il l’aurait construit à partir de l’épine dorsale d’un virus connu pour causer des maladies. Mais les scientifiques ont découvert que l’épine dorsale du SRAS-CoV-2 différait considérablement de celle des coronavirus déjà connus et ressemblait principalement à des virus apparentés trouvés chez les chauves-souris et les pangolins.
"Ces deux caractéristiques du virus, des mutations dans la partie RBD de la protéine Spike et son squelette distinct, excluent toute manipulation en laboratoire comme origine possible du SRAS-CoV-2", a déclaré Andersen.
Josie Golding, PhD, responsable de l’épidémie au Wellcome Trust, basé au Royaume-Uni, a déclaré que les découvertes d’Andersen et de ses collègues sont "d’une importance cruciale pour fournir une vision fondée sur des preuves des rumeurs qui circulent sur les origines". du virus (SARS-CoV)-2) causant le COVID-19.
"Ils concluent que le virus est un produit de l’évolution naturelle", ajoute Goulding, "mettant ainsi fin à toute spéculation sur une ingénierie génétique délibérée".
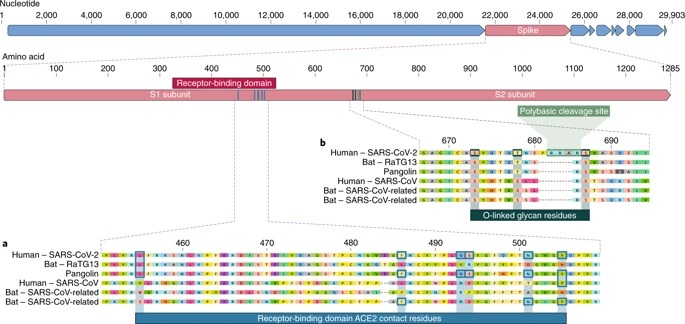
a, Mutations dans les résidus de contact de la protéine de pointe du SRAS-CoV-2. La protéine de pointe du SRAS-CoV-2 (barre rouge en haut) a été alignée sur les coronavirus de type SRAS-CoV plus étroitement liés et sur le SRAS-CoV lui-même. Les résidus clés sur la protéine de pointe qui entrent en contact avec le récepteur ACE2 sont marqués par des cases bleues dans le SRAS-CoV-2 et les virus apparentés, y compris le SRAS-CoV (souche Urbani). b, Acquisition du site de clivage polybasique et des glycanes liés à l’O. Le site de clivage polybasique et les trois glycanes O-liés prévus adjacents sont uniques au SRAS-CoV-2 et n’ont pas été observés auparavant dans les bêtacoronavirus de la lignée B. Les séquences présentées proviennent de NCBI GenBank, codes d’accès MN908947, MN996532, AY278741, KY417146 et MK211376. Les séquences du coronavirus pangolin sont un consensus généré à partir de SRR10168377 et SRR10168378 (NCBI BioProject PRJNA573298) 29,30.
Origines possibles du virus
Sur la base de leur analyse de séquençage génomique, Andersen et ses collaborateurs ont conclu que les origines les plus probables du SRAS-CoV-2 suivaient l’un des deux scénarios possibles.
Dans un scénario, le virus a évolué jusqu’à son état pathogène actuel par sélection naturelle chez un hôte non humain, puis s’est transmis aux humains . C’est ainsi que de précédentes épidémies de coronavirus sont apparues, des humains contractant le virus après une exposition directe à des civettes (SRAS) et des chameaux (MERS). Les chercheurs ont proposé les chauves-souris comme réservoir le plus probable du SRAS-CoV-2, car elles sont très similaires au coronavirus des chauves-souris. Cependant, il n’existe aucun cas documenté de transmission directe entre chauves-souris et humains, ce qui suggère qu’un hôte intermédiaire était probablement impliqué entre les chauves-souris et les humains.
Dans ce scénario, les deux caractéristiques distinctives de la protéine Spike du SRAS-CoV-2, la partie RBD qui se lie aux cellules et le site de clivage qui ouvre le virus, auraient évolué vers leur état actuel avant de pénétrer chez l’homme. . Dans ce cas, l’épidémie actuelle serait probablement apparue rapidement dès que les humains auraient été infectés, car le virus aurait déjà développé les caractéristiques qui le rendent pathogène et capable de se propager entre les personnes.
Dans l’autre scénario proposé, une version non pathogène du virus est passée d’un hôte animal à l’homme, puis a évolué vers son état pathogène actuel au sein de la population humaine. Par exemple, certains coronavirus des pangolins, des mammifères ressemblant à des tatous trouvés en Asie et en Afrique, ont une structure RBD très similaire à celle du SRAS-CoV-2. Un coronavirus provenant d’un pangolin aurait pu être transmis à un humain, soit directement, soit par l’intermédiaire d’un hôte intermédiaire, comme des civettes ou des furets.
Ainsi, l’autre protéine de pointe caractéristique du SRAS-CoV-2, le site de clivage, pourrait avoir évolué au sein d’un hôte humain, éventuellement grâce à une circulation limitée non détectée dans la population humaine avant le début de l’épidémie.
Les chercheurs ont découvert que le site de clivage du SRAS-CoV-2 semble similaire aux sites de clivage des souches de grippe aviaire qui se propagent facilement entre les personnes. Le SRAS-CoV-2 aurait pu développer un site de clivage aussi virulent dans les cellules humaines et déclencher rapidement l’épidémie actuelle, car le coronavirus serait peut-être devenu beaucoup plus capable de se propager entre les personnes.
Le co-auteur de l’étude, Andrew Rambaut, a averti qu’il est difficile, voire impossible, de savoir à ce stade quel scénario est le plus probable. Si le SRAS-CoV-2 pénétrait chez l’homme sous sa forme pathogène actuelle à partir d’une source animale, la probabilité de futures épidémies augmenterait, car la souche du virus à l’origine de la maladie pourrait encore circuler dans la population animale et réapparaître. sauter les humains . Les chances sont moindres qu’un coronavirus non pathogène pénètre dans la population humaine et développe ensuite des propriétés similaires à celles du SRAS-CoV-2.















